cryoEM Refinement Tools
Download software
Click here to download cryoEM Refinement tools.
System requirements
Linux operation system with sufficient memory (e.g. >1G) and hard disk (e.g >10G) to support cryoEM map comparison and storage of a large number of conformations generated during refinement.
Building and refining atomic models within cryoEM maps
The methods described in this page were developed in a close collaboration with the Electron Imaging Center for Nanomachines (EICN) at UCLA directed by Dr. Z. Hong Zhou. Our long-term goal is to develop a comprehensive computational platform for deriving atomic models from the cryo-electron microscopy (cryoEM') maps of macromolecular complexes.
The multi-scale refinement approach used in the construction of the grass carp reovirus (GCRV) model involves two consecutive stages: a modeling-based protocol that uses EM-IMO as its core component to refine protein models within cryoEM maps and an MD-based final refinement aimed to adjust structural details using energy and forces derived from a grid representation of cryoEM maps. The details about the EM-IMO method, the modeling-based protocol and our version of MDFF method are provided below.
1. EM-IMO
Description: EM-IMO was based on the framework of IMO procedure (Zhu J et al, Proteins 2006), which was originally developed for local structure refinement in the context of protein structure prediction. The difference between IMO and other local modeling programs such as Loopy (Xiang et al, PNAS 2002) is that IMO allows secondary structure elements to be treated as rigid bodies and to be rebuilt and modified during refinement. In another word, IMO was developed primarily for modeling and refining secondary structure elements, although it is equally applicable to loop regions, in which the number of SSEs is simply zero. The rationale behind EM-IMO is that after fitting a protein model into the cryoEM map the discrepancies between the model and the map are often localized to certain regions of the structure and can be readily recognized through visual inspection. Thus, a method such as EM-IMO that can refine protein structure locally using cryoEM map as a constraint will be very useful in the model building process in the cryoEM study of macromolecular complexes. A flow-chart of EM-IMO procedure is shown below.

Implementation: Similar to IMO, the current implementation of EM-IMO combines a master script (em_imo.pl) and several modular programs written in C or C++. This design allows us to easily test various combinations of modules and to incorporate new functions with the least change to the original structure. For example, the scoring function in IMO has been extended in EM-IMO by adding a new module, which is used to calculate the density fitting score for each candidate conformation during sampling. Another example is that the module used to calculate DFIRE energy in IMO is now replaced with a new module used to calculate normalized DFIRE energy in EM-IMO. The only problem of such implementation is the low speed, which will be addressed in the further development. To be more specific, all the modules will be integrated into a single data structure and more efficient methods will be used for side chain packing and density fitting. Our preliminary results indicated that such change could improve the speed by a factor of 20 or even more.
Installation: Please follow the instruction below to install the EM-IMO refinement program.
Step 1: Download the EM-IMO package and unpack it in your home directory (e.g. /home/Jane_Doe/) *** NOTE: THIS LINK DOESN'T WORK ***
Step 2: Download the SCRWL3.0 package and EMAN (version 1.9) package and install them in your home directory.
Step 3: Copy the SCRWL executable to the bin directory in the EM-IMO directory (e.g. /home/Jane_Doe/em_imo/bin).
Step 4: Compile "quickccc.cpp" in the same bin directory as above using g++ and following options:
g++ -O3 -o quickccc quickccc.cpp -IXXX/EMAN/include -LXXX/EMAN/lib -1EM
(XXX: path that specifies where the EMAN package is located)
Step 5: Compile "normdf151_cmx.cpp" in the same bin directory as above using g++ and following options:
g++ -O3 -o normdf151_cmx normdf151_cmx.cpp
Step 6: Set up the environmental variable - EM_IMO_DIR, (e.g. in .cshrc if you are using C-Shell)
setenv EM_IMO_DIR /home/Jane_Doe/em-imo
Step 7: Create a working directory and copy files for the region to be refined (e.g. for a protein "abc")
mkdir /home/Jane_Doe/abc/region_1
cd /home/Jane_Doe/abc/region_1
cp /home/Jane_Doe/em_imo/input/input.prm . (other parameter fiels too if needed)
Step 8: Change the parameters in the input file and parameter files accordingly
Step 9: Perform EM-IMO refinement by running the master script:
/home/Jane_Doe/em_imo/script/em_imo.pl -i input.prm
Step 10: The final output - a PDB-format file - will be in the same directory along with other intermediate output files.
2. Modeling-based refinement protocol
Description: By design, EM-IMO is a local refinement program that can only refine one region at a time. If there exist multiple regions that are inconsistent with the cryoEM map, we can refine them simultaneously and then combine the refined structures into a final model. After energy minimization, the refined model can be re-fitted into the cryoEM map and used as the initial model for the next iteration of refinement. This idea has been implemented in the "modeling-based refinement protocol" and applied to the construction of GCRV model within a near-atomic resolution cryoEM map. By refining multiple regions in an iterative manner, this protocol is actually improving the fit within the cryoEM map at a global level. A flow-chart of the modeling-based protocol is shown below.
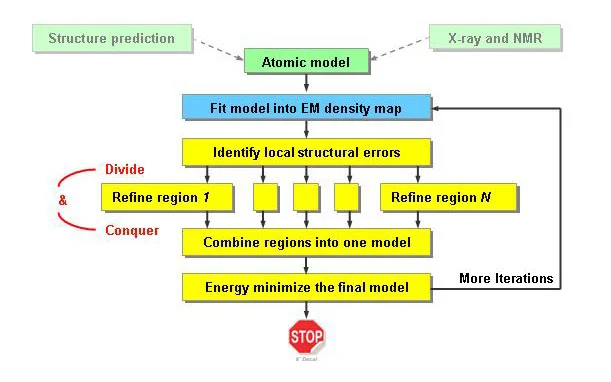
Implementation: The fitting of an atomic model into the cryoEM map can be done using UCSF Chimera. Two graphic programs, UCSF Chimera and GRASP2, can be used together to identify the regions that are inconsistent with the cryoEM map. Specifically, UCSF Chimera can be used to compare the density map and the structure, while GRASP2 can be used to check sequence alignment, compare secondary structure assignment and prediction, and compare protein structures after structural alignment. The local refinement using cryoEM maps as a constraint can be done with the EM-IMO method. After refined regions are merged into a single model, an energy minimization can be performed to further relax the structure using any molecular mechanics program such as TINKER.
3. In-house implementation of MDFF
Description: Molecular Dynamics Flexible Fitting (MDFF) was originally developed by Schulten and co-workers (Trabuco et al, Structure 2008) and used in the refinement of ribosome structure within a 6.7Å-resolution cryoEM map. In our study, we re-implemented the MDFF method in the GROMOS96 MD package and used it in the end-stage refinement of GCRV proteins. In our implementation, the GROMOS96 43a1 force filed was used in conjunction with the mAGB implicit solvation model and a cryoEM term that derives energy and forces from the grid representation of cryoEM map using linear interpolation. Currently we are testing other implicit solvent models in order to speed up the MD simulation without losing the solvation effect, which may be crucial for structure refinement.
Implementation: The MDFF method was implemented in Fortran 90 and can be downloaded here***LINK DOESN'T WORK*** as a single file. You need to add a functional call to reademgrid in both runem.f and runmd.f, and then a functional call to gridforce in forces.f. If you are really interested in using this version of MDFF, please get a licensed version of GROMOS96 MD package and send me an email to ask for all the source code files and corresponding Makefile.
References
Zhu J, Cheng L, Fang Q, Zhou ZH, Honig B. Building and refining protein models within cryo-electron microscopy density maps based on homology modeling and multiscale structure refinement. J Mol Biol. 2010 Apr 2;397(3):835-51.
Acknowledgments
cryoEM Refinement Tools is supported by funding from the National Institutes of Health Grant #GM30518 and the National Science Foundation Grant #MCB-0416708.
Developed in the Honig Lab
Questions
If you have any questions regarding the cryoEM refinement programs, please contact Dr. Jiang Zhu.